






Symptoms
What Are the Symptoms of Congenital Sucrase-Isomaltase Deficiency?
Symptoms in individuals with Congenital Sucrase-Isomaltase Deficiency (CSID) can range from mild to severe gastrointestinal (GI) complaints such as chronic, watery, acidic diarrhea; gas; bloating; and abdominal pain.
In This Section
Symptoms of CSID may not be apparent in infants until they begin to eat foods that contain sucrose and starch-containing foods (for example, juices, solid foods, medications sweetened with sucrose). This is because breast milk does not contain sucrose.1
Chronic, watery diarrhea and failure to thrive are the most common symptoms in infants and toddlers.1 Other symptoms include abdominal distention (swelling), gassiness, colic, irritability, scratched and reddened buttocks, severe diaper rash due to acidic diarrhea, indigestion (dyspepsia), and vomiting.1-3 In some ethnic groups, notably Greenland Eskimos and some Alaskan natives, the GI symptoms associated with CSID may not be as prominent if they eat a diet traditional in their community that is low in carbohydrates and high in proteins and fats.1,2
A small number of patients affected by CSID may need to be hospitalized for diarrhea-induced dehydration, malnutrition, muscle wasting, and weakness.1-3 Patients with a confirmed diagnosis of CSID commonly report being examined first by their doctors for more common GI disorders, including toddler’s diarrhea, irritable bowel syndrome with diarrhea (IBS-D), celiac disease, cystic fibrosis, and food allergies.1,4
Congenital Sucrase-Isomaltase Deficiency is not a disease that a patient can outgrow.
Indeed, symptoms persist in adults. However, GI symptoms associated with CSID can vary. For example, GI symptoms experienced by adults may not be as severe as the GI symptoms experienced by children.1
In some adults, the symptoms may be limited to an increase in bowel movement (BM) frequency, reduced stool consistency (looser stools or watery stools), abdominal distention (swelling), and flatulence (gas). Episodic watery diarrhea may also occur after eating a meal that contains high levels of sucrose. In some individuals affected with CSID, diarrhea may alternate with constipation, particularly when taking common antidiarrheal medications, which may lead to a misdiagnosis of another GI condition, such as alternating or mixed-type irritable bowel syndrome (IBS-A).1
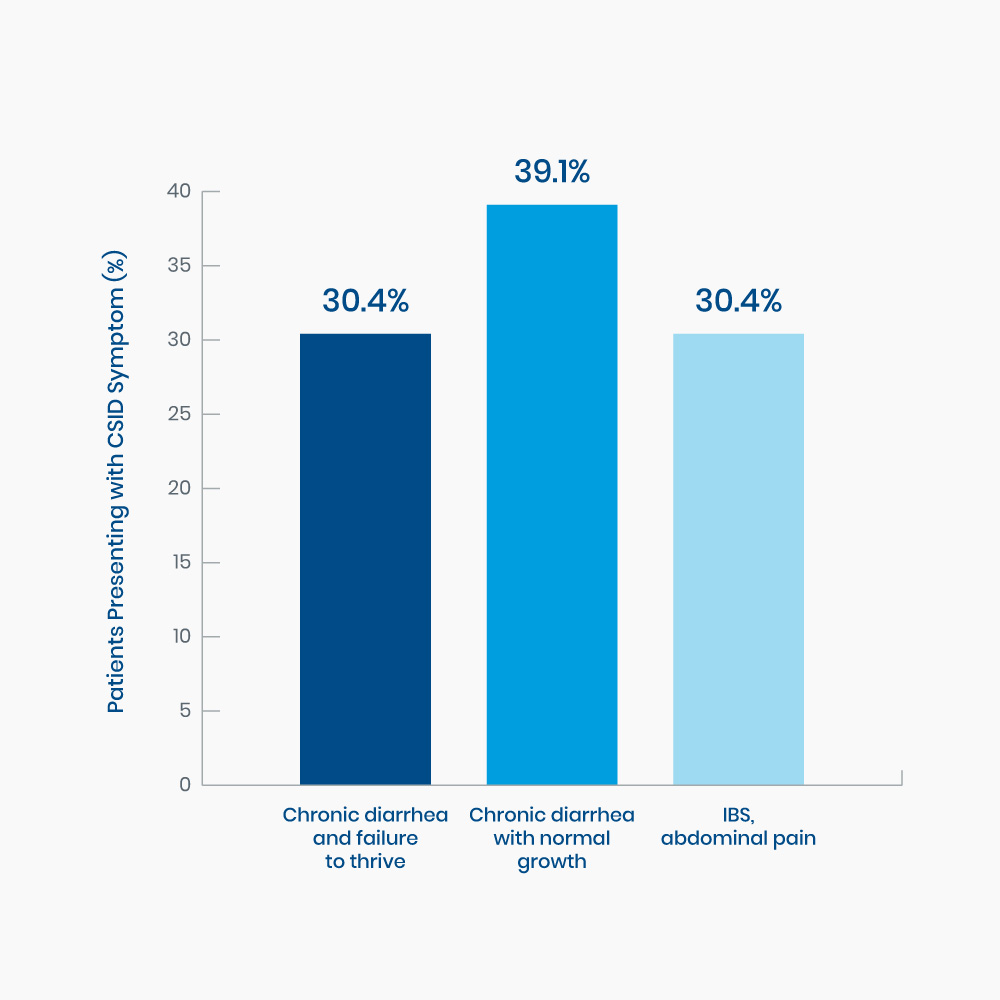
Figure 1. Range of GI symptoms present in twenty-three children when diagnosed with CSID.1
Symptoms Consistent with a Diagnosis of CSID
Table 1. Overview of CSID Symptoms
Common Signs and Symptoms
Individuals with Congenital Sucrase-Isomaltase Deficiency (CSID) typically have one or more of the following symptoms:
- Chronic diarrhea and/or loose stools (more severe or explosive in young children or babies after they have stopped breastfeeding)
- High frequency of bowel movements (BMs)
- Reports of particularly foul BM odor related to poor absorption of carbohydrates
- Gas and abdominal bloating
- Abdominal pain
- Complaints of nausea or indigestion
- Diarrhea mixed with intermittent constipation, particularly when chronically taking common drugs to stop diarrhea
- Onset of symptoms soon after consuming a meal
- A low body mass index (BMI) that falls below the age-appropriate growth chart curve or failure to thrive when very young
- Avoidance or intolerance of foods containing carbohydrates, particularly sugary sweets or starches (for example, potatoes, rice, pasta)
- Lack of relief from treatment with common drugs to stop diarrhea
- Long history of examinations by multiple gastroenterologists for unusual GI conditions with symptoms similar to CSID, such as inflammation of the gall bladder (cholecystitis); intolerance of foods containing gluten (proteins found in grains), called celiac disease; an inherited disorder that impairs the secretion of mucus, called cystic fibrosis; and impaired absorption of bile acid (naturally-occurring molecule that helps the body break down fats); all these conditions also may cause chronic diarrhea
Family History of GI Symptoms
Since CSID is an inherited disorder, other family members may also be affected:
- Parents and/or siblings may have similar GI symptoms after a meal
- They may avoid eating carbohydrates and sweet foods
Symptom Frequency and Duration
- Individuals with CSID may experience GI symptoms frequently (several times a week or every day)
- Onset of symptoms is shortly after eating a meal
- Symptom have been a problem for a very long-time (since childhood or for many years); in children, symptoms may start when no longer breastfeeding, with increased consumption of dietary carbohydrates in baby foods
References
- Treem WR. Congenital Sucrase-Isomaltase Deficiency. J Pediatr Gastroenterol Nutr. 1995;21(1):1-14. doi:10.1097/00005176-199507000-00001
- Gudmand-Høyer E. Sucrose malabsorption in children: a report of thirty-one Greenlanders. J Pediatr Gastroenterol Nutr. 1985;4(6):873-877. doi:10.1097/00005176-198512000-00005
- Antonowicz I, Lloyd-Still JD, Khaw KT, Shwachman H. Congenital Sucrase-Isomaltase Deficiency. Observations over a period of 6 years. Pediatrics. 1972;49(6):847-853.
- Treem WR. Clinical aspects and treatment of Congenital Sucrase-Isomaltase Deficiency. J Pediatr Gastroenterol Nutr. 2012;55(suppl 2):S7-S13. doi:10.1097/01.mpg.0000421401.57633.9